

Hi! My name is Jorge Suárez Recio, I am from Oviedo, Spain, and I am going to explain to you an internship at the Fusion Group at the Barcelona Supercomputing Center (BSC) that I completed this summer from July 1 to August 30.
About me
I graduated in Physics from the Universidad de Oviedo (UO), where I later completed a master´s degree in Nanotechnology and Advanced Materials. It was during this period that I became interested in computational science, and more specifically, in nuclear materials-related physics. In September 2021, I started a Ph.D. at the Universidad Politécnica de Madrid (UPM) on the characterization and modeling of new materials for inertial and magnetic fusion. During this first year, I’ve been focused on Density Functional Theory (DFT) simulations, specifically using the Vienna Ab initio Simulation Package (VASP), to study plasma-facing materials (PFM) in future nuclear fusion reactors.
The main reason I was interested in doing this virtual internship and collaborating with José Julio Gutiérrez Moreno was to learn from his experience related to the computer software called Large-scale Atomic/Molecular Massively Parallel Simulator (LAMMPS), an open-source Molecular Dynamics (MD) code.
Motivation
One of the most demanding challenges confronted by the construction of commercial nuclear fusion reactors is the development of new advanced materials that are more resistant to extreme operating conditions. Tungsten is postulated as one of the most promising candidates as a plasma-facing material (PFM) in future fusion reactors, as it satisfies most of the highly demanding requirements. However, prior work indicates that W also has major drawbacks, a key one being its tendency to easily retain light species (LIA, light interatomic atoms), which causes the formation of bubbles, cracks, and sputtering of the material. Recently, numerous publications have reported that nanostructured materials are more resistant to radiation, mainly due to the high density of grain boundaries (GB). This implies that the overpressurization of the LIA bubbles can be delayed, thus increasing the limit of radiation resistance that the material can endure without seriously compromising its performance. In an optimal scenario, the GBs would not only behave as defect sinks but as effective diffusion channels as well, by promoting the outgassing of defects.
Due to the high computational cost of DFT calculations when dealing with complex arrangements such as the ones tackled here, a promising approach is to introduce MD simulations in our multiscale methodology, since this type of calculation is much less computationally expensive albeit less accurate. Once the systems are relaxed with MD, the initial DFT configurations become already closer to equilibrium and consequently, the expected computational time is greatly reduced and, while simultaneously allowing for the testing of the MD interatomic potential.
Project
During my first weeks, I gained basic knowledge of LAMMPS thanks to some scripts Julio provided me. Once I got the fundamental skills, we focused on calculating migration barriers. These calculations were carried out within the Transition State Theory (TST) framework, as implemented in the Nudged Elastic Band (NEB) method. The NEB is a method for finding saddle points and minimum energy paths. The method works by optimizing a number of intermediate images along the reaction path, where each of the intermediate images is run simultaneously in parallel. E. g. if there are 10 replicas, the second image will assign a position that is 10% of the distance along a line between the starting and final point, and the 9th image will assign a position that is 90% of the distance along the line. Each image finds the lowest energy possible while maintaining equal spacing to neighboring images. This constrained optimization is done by adding spring forces along the band between images and by projecting out the component of the force due to the potential perpendicular to the band.
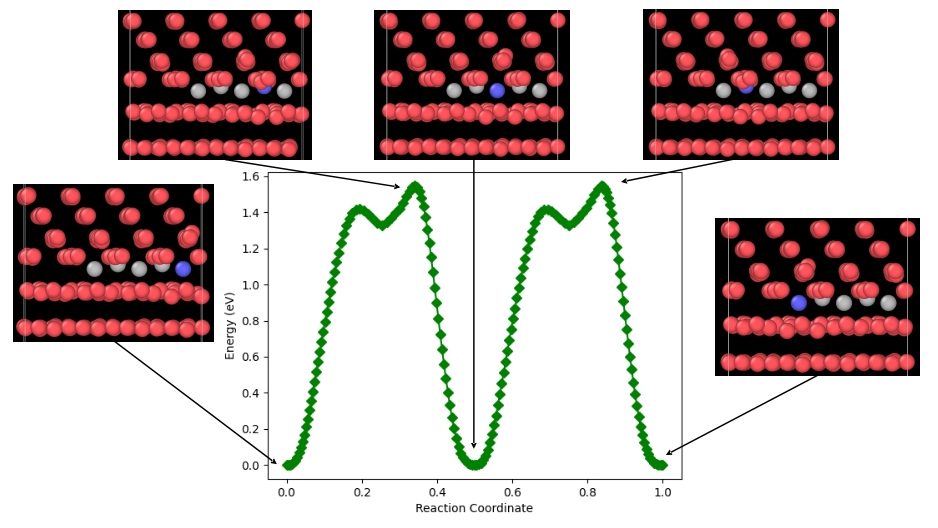
The learning curve needed to garner expertise in this type of calculation took me most of the time. Firstly, I calculated migration barriers in W bulk to easily compare the values I obtained to DFT and experimental, and thus test the interatomic potential. Once the potential was validated, I began to study migrations both of W interstitials and He on a W(110) – W(112) GB (456 atoms) interface. Fig. 1 shows the migration of a SIA atom along the Y-axis through the GB. All these configurations were run on the MareNostrum Supercomputer at BSC. Thanks to this, I could run many more images simultaneously in a shorter time, a win-win!
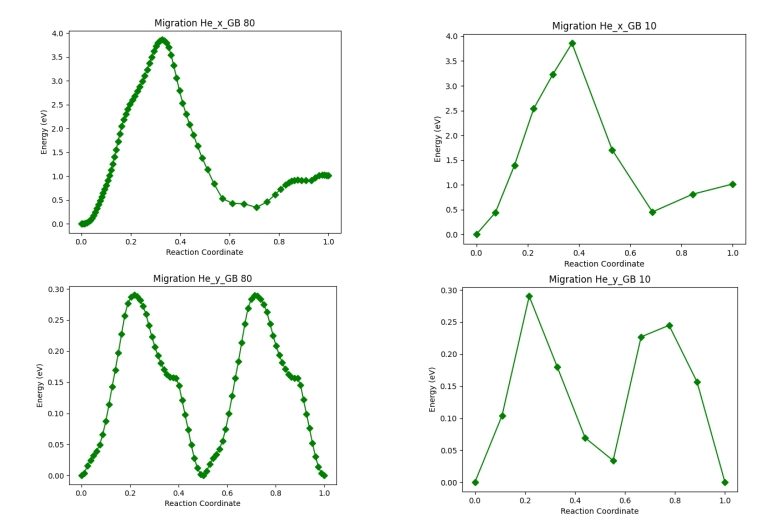
As the main idea was to run these migrations later with VASP and I was using 160 images in LAMMPS, something impossible to tackle with DFT, I sequentially reduced the number of images of a few of the most interesting barriers to see if the shape and, mainly, the energy of the barrier was maintained. We can see in Fig. 2 that the results were quite good.
Moreover, I have been testing a couple of interatomic potentials in the presence of vacancies, di vacancies, self-interstitial atoms (SIA), and more. Basically, this meant a re-relaxation through the MD potentials, taking as a starting point the previously relaxed DFT configurations of the W(110)/W(112) GB with defects, relaxing it again with the MD interatomic potential, and studying how much the atoms in the system moved. This allowed us to quantify the reliability of the potentials at the interface with defects. The goal of this potential testing is to use them later for Quantum Mechanics / Molecular Mechanics (QM/MM) simulations. The hybrid QM/MM approach is a molecular simulation method that combines both the strengths of ab initio QM calculations (accuracy) and MM (speed) approaches.
Experience
Even though my stay was held virtually due to the restrictions imposed by the Covid pandemic, Julio Gutierrez was always there for me whenever I needed him. Every time I had a question, he answered it as soon as possible, usually through Skype chat, but also, if necessary, we made a video call, to discuss or share the screen. Last but not least, I’d like to express my gratitude to Mary Kate Chessey, Arda Erbasan and Dominik Freinberger for their help and efforts to make me feel like a part of the team. Also, thanks to my supervisors, Raquel González Arrabal and Roberto Iglesias Pastrana, for bringing me this opportunity.
My research was funded by the Spanish Ministry of Science and Innovation, through the projects RADIAFUS V, grant number PID2019-105325RB-C32 and predoctoral grant number PRE2020-096178. It was also funded by the Foundation for the Promotion in Asturias of Applied Scientific Research and Technology (FICYT): AYUD/2021/51822 (GRUPIN—MAGNES).
The research carried out by the BSC Fusion group was partly funded by the FusionCAT project (001-P-001722) which has been 50% co-financed with € 1.960.963,66 by the European Fund for Regional Development of the European Union within the framework of the 2014-2020 ERDF Operational Program of Catalonia, with the support of the Generalitat of Catalonia.
