

On 27 June, group members Julio Gutiérrez and Mary Kate Chessey attended a Materials Square webinar (#118) titled, “Machine learning interatomic potentials for modelling radiation damage,” with invited speaker Professor Kai Nordlund of Computational Materials Physics from the University of Helsinki, Finland.
Here is the abstract of the talk:
The modern theoretical understanding of the effects of ionizing radiation on materials – which can be both beneficial or detrimental – relies very much on atom-level simulations. Their reliability in turn depends crucially on the reliability of the interatomic interaction model. In this talk I will present recent developments of machine-learning interaction models suitable for radiation damage calculations for bcc high-entropy alloys and fcc metals, that are much more accurate than the traditional analytical interatomic potentials.
Major improvements in the accuracy of the latest interatomic potentials for the kinds of defects associated with high-energy neutron irradiation of tungsten stem primarily from the use of training data that includes the local energies of close range interactions between atoms (less than one tenth of one nanometer apart, or interaction energies above 10 eV). The training data was enhanced using calculations of energies of a variety of defects and liquid configurations of atoms. These close range interactions are a crucial aspect of fusion reactor materials modelling due to the impacts of high energy (14.1 MeV) neutrons released from the fusion plasma, which irradiate the first wall components.
In our Fusion Group at the Barcelona Supercomputing Center,we found this webinar very interesting. As part of the FusionCAT project, we study similar materials and defects (irradiated tungsten) using multiple approaches: classical molecular dynamics using LAMMPS; ab-initio density functional theory (DFT) using BigDFT; and development of machine learning interatomic potentials using QuantumATK. Classical molecular dynamics relies on fast, accurate interatomic potentials to scale up system sizes and timescales compared to what is computationally feasible using costly ab-initio methods. Highly accurate DFT calculations form training data for machine learning interatomic potentials. Finally, machine learning approaches optimize between the accuracy of ab-initio methods and the speed needed for larger system sizes modelled using classical molecular dynamics methods.
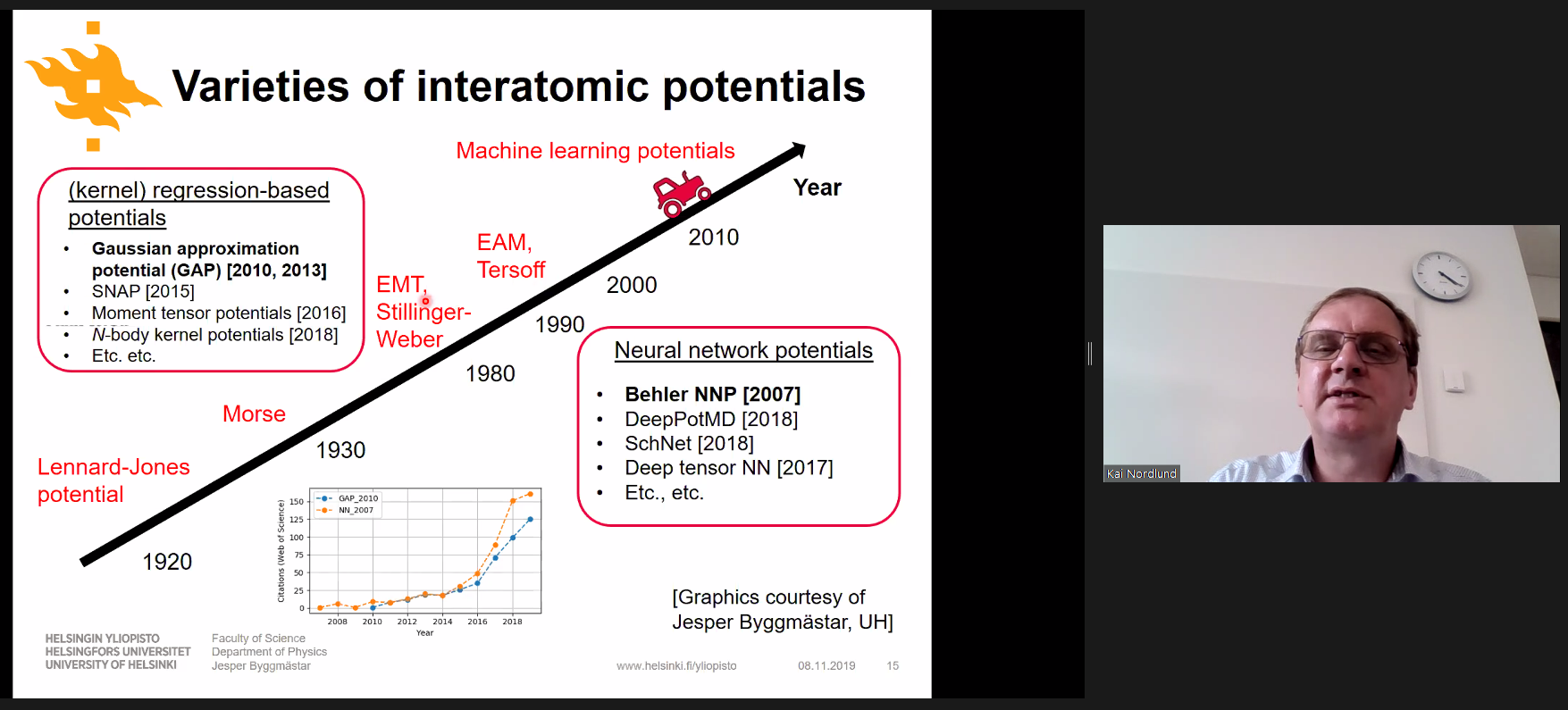
During the question and answer session, Professor Nordlund stated that a full Gaussian Approximation Potential (GAP) machine learning potential can be at worst five to ten orders of magnitude slower to calculate than an Embedded Atom Model (EAM) potential due to the multiple levels of machine learning training used by GAP. Fortunately, current research in this area focuses on ways of making machine learning potentials faster without sacrificing too much accuracy! See Jesper Byggmästar’s work on tabGAP for more details.

If you’re interested in a fully-funded four year PhD position in a productive lab composed of a mixture of experimentalists and simulation researchers, send an email to Kai Nordlund! One PhD will focus on modelling high-flux ion implantation in silicon and the other on modelling metals used in fusion reactor materials.

Attendees enjoyed this picture taken from Vartiokylänlahti Bay of the “white nights” of Helsinki, Finland in the summer, where the sky is bright even until 23h!
The FusionCAT project with reference number 001-P-001722 has been 50% co-financed with € 1.960.963,66 by the European Fund for Regional Development of the European Union within the framework of the 2014-2020 ERDF Operational Program of Catalonia, with the support of the Generalitat of Catalonia.
